Elsayed Hegazy1, Mahmoud Elhefnawi1, 2
1Nile University, Giza, 12588, Egypt.
2National Research Centre, Cairo, Egypt.
Abstract
As next-generation sequencing costs continue to fall and personalized medicine moves closer to routine clinical implementation, there is growing demand among researchers and clinicians for accessible web-based tools that can translate raw variant call format (VCF) data into clinically actionable phenotype information. Personalized medicine and the highly attention of next generation sequencing increase the demand of turning the genotype data into meaningful phenotype data. VarPhen is a web based tool used to do such thing. It’s written in C# code it’s based on using RefSeq SNPs ID as a genotype to retrieve the relevant phenotype. VarPhen use ClinVar database as the source of clinical information and phenotypes relevant to specific variant. Students and researchers studying bioinformatics, genomic medicine, or clinical genetics will find tools like VarPhen particularly relevant as they illustrate how public databases such as ClinVar can be leveraged to build no-registration, no-cost phenotype interpretation services accessible to non-programming users.
Introduction
Next generation sequencing workflows and pipelines is now available for analyze all row data from quality control and mapping to variant calling but very few tools deals with vcf file to interpret to generate a meaningful reports with the common and rare diseases. The gap between high-throughput variant detection and clinically meaningful phenotype interpretation remains one of the central bottlenecks in translational genomics, particularly for laboratories operating without dedicated bioinformatics staff. One of the biggest servers regarding this issue is ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) which provides a freely available archive of the relationships among medically important variants and phenotypes. ClinVar is a huge database for reporting human variation, interpretations of the relationship of that variation to human health and the evidence supporting each interpretation. The database is tightly coupled with dbSNP and dbVar, which maintain information about the location of variation on human assemblies. ClinVar is also based on the phenotypic descriptions maintained in MedGen (http://www.ncbi.nlm.nih.gov/medgen). Each ClinVar record represents the submitter, the variation and the phenotype. As of 2024, ClinVar contains interpretations for more than one million variants from thousands of submitters worldwide, making it the most widely referenced variant pathogenicity repository for clinical genetics workflows (Landrum et al., 2018).
The demand of vcf interpretation to valuable knowledge and phenotype increased day by day with the increasing of personal genome demand day by day.
Here we will develop web based application that able to connect to CinVar and retrieve diseases associated with each variant listed in vcf file or sample.
Availability and implementation
VarPhen available for use on http://www.varphen.com as a web based tool written in ASP.Net with C# code behind using NCBI Database ClinVar API as a source of phenotypes. The platform requires no local software installation and no user registration, lowering the barrier to entry for clinical geneticists, medical students, and biomedical researchers who need rapid phenotype screening from small variant files.
Review of literature
Writing a Similar Assignment?
Get a Scholar-Written Paper Matched to Your Brief
Every order is handled by a degree-holding expert in your subject — written to your exact rubric, fully original, and delivered ahead of your deadline.
Start My OrderKnowledge is more valuable when shared. Collaborative data sharing in genomics has been shown to accelerate the reclassification of variants of uncertain significance, which in turn may directly improve diagnostic yields in rare disease clinics (Harrison et al., 2023). By contributing these tools to the big spectrum which is the research community and healthcare as industry, we want to increase the quality and accuracy of genetic data analysis and interpretation available to all patients, physicians and researchers.
OpenSNP is a Crowdsourced Web Resource for Personal Genomics. It’s based on collecting users or patient’s vcf files from different sources like 23andme and decodeme plus the normal vcf file then detect variants and all relevant phenotypes.
CLINVITAE is a clinically observation database uses the genetic variants aggregated from public sources. It is operated and made freely available by INVITAE which is a service like ClinVar.
To make CLINVITAE as informative as possible, CLINVITAE aggregate the data from multiple public databases. CLINVITAE long term goal is to facilitate the search for clinically interpreted variants by creating a single unified resource for all interpretation results. CLINVITAE want physicians and researchers to save their time when comparing variants across multiple platforms and resources, and fully utilize the available data.
GWAS Central or the Human Genome Variation database of Genotype-to-Phenotype information which is a database of summary level findings from genetic association studies, both large and small. GWAS actively gather datasets from public domain projects, and encourage direct data submission from the community improving the quality and accuracy of interpretation.
Genome-wide association studies (GWAS) have been successful at identifying some of the variation in traits attributable to genetics. The National Human Genome Research Institute (NHGRI) has begun aggregating results of association studies into a master GWAS catalog. The NHGRI-EBI GWAS Catalog, as of 2024, catalogs more than 500,000 unique variant-trait associations from over 6,700 publications, representing a substantial expansion from its initial scope and confirming the database’s central role in human complex-trait genetics research.
Also, INTERPRETOME is a freely available and secure personal genome interpretation engine analyze vcf file into valuable knowledge for diseases from GWAS.
The Human Gene Mutation Database from QIAGEN represents a longstanding effort to collect known published gene lesions responsible for human inherited disease.
Also, The Diagnostic Mutation Database (DMuDB) is a secure repository of clinical quality variant data collected from diagnostic genetics laboratories. Access to DMuDB is available by annual laboratory subscription, and must be for diagnostic purposes only.
Many of databases and tools do such analysis or job but very few tools and databases are freely available or accessible by programming inside your application. A key limitation of most existing tools is their dependency on local installation, programming competency, or institutional subscriptions, all of which exclude a large segment of potential users in low- and middle-income research settings.
Aim
Developing web based application for transforming variants from vcf into knowledge by identifying which variant pathogenic and what is the associated diseases with that variant.
Stuck on Your Assignment?
Cola Papers Experts Are Ready Right Now
Join thousands of students who submit confidently. Human-written, plagiarism-checked, and formatted to your institution's exact standards.
Methods
Technically this web based tool developed straight forward by using one of the most powerful web technologies which is ASP.Net web forms with C Sharp back end code. User asked to upload vcf file then file processing done by manipulating file to discard vcf header and start reading vcf data after the header. After that VarPhen only read the third column which represent the RefSeq of the variant as ClinVar input. Also VarPhen detect if the SNP is novel or not this improve the tool performance because if VarPhen found a novel variant so there no web request created to ClinVar but if the variant not novel so VarPhen create a web request to ClinVar asking for full listed information associated with this variant. After the web request processed ClinVar web response retrieved by VarPhen as XML file. VarPhen start to parse and analyze the resulted XML file. Manipulation of XML file target is extracting the phenotypes which associated with the variant of interest. VarPhen uses C# data structure Queue which help also to improve the performance of the VarPhen web requests to ClinVar API because it’s based on the concept of First in first served first out. All phenotypes saved in another C# data structure which is the List data structure as a series of strings. After that it’s the turn of the presentation layer by populating ASP.Net grid view by the list of phenotypes as a table in the user interface. The queue-based architecture is particularly advantageous when processing large VCF files with hundreds of known variants, as it prevents server overload by serialising API calls to ClinVar rather than launching simultaneous requests.
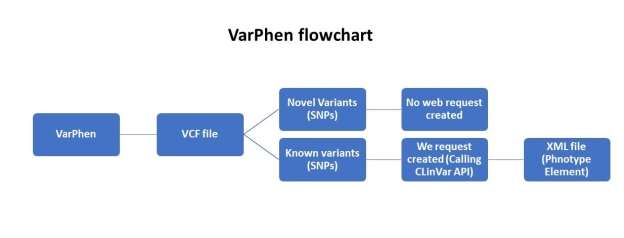
Figure 1- VarPhen flowchart
Figure 1 shows the flowchart of VarPhen tool as it indicated its start with vcf file the check if variant is novel or not. No web request created in case of variant novel. API calling starts only with known variants to retrieve the XML file containing the phenotype data.
Conclusion
VarPhen is one of the easiest ways to know what phenotypes associated with a specific vcf file is. VarPhen target users with no programming experience. No registration required to use it. So simple by its user friendly interface. Future development could include support for HGVS nomenclature inputs, batch processing capabilities, and integration with additional databases such as ClinGen and OMIM to expand the scope and accuracy of variant-to-phenotype mapping available to clinical and research users alike.
Web-based genotype-to-phenotype tools such as VarPhen occupy an increasingly important niche in the genomic medicine ecosystem, particularly as whole-genome and whole-exome sequencing become more accessible in clinical diagnostics. The challenge of variant interpretation — moving from a list of genomic differences to a ranked set of clinically relevant phenotypic associations — remains computationally and informatically demanding for laboratories without specialist support. Tools that leverage well-curated public variant databases through API calls, without requiring users to install software or manage dependencies, may play a significant role in democratising genomic literacy among medical students, allied health professionals, and researchers in resource-limited settings. Landrum et al. (2018) documented ClinVar’s trajectory toward becoming a more granular, evidence-tiered resource, with submitter-specific interpretations increasingly subject to expert panel review, which suggests that API-reliant tools like VarPhen will continue to benefit from the ongoing curation efforts of the broader clinical genetics community. Ensuring that such tools are regularly updated to reflect ClinVar’s evolving data standards and submission guidelines will be critical for maintaining the accuracy and clinical relevance of phenotype outputs.
References
Harrison, S. M., Biesecker, L. G., & Rehm, H. L. (2023). Overview of specifications to the ACMG/AMP variant interpretation guidelines. Current Protocols, 3(3), e705. https://doi.org/10.1002/cpz1.705
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., & Maglott, D. R. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. https://doi.org/10.1093/nar/gky1162
Manolio, T. A., Rowley, R., Williams, M. S., Roden, D., Ginsburg, G. S., Bult, C., & Green, E. D. (2019). Opportunities, resources, and techniques for implementing genomics in clinical care. The Lancet, 394(10197), 511–520. https://doi.org/10.1016/S0140-6736(19)31140-7
Rehm, H. L., Berg, J. S., Brooks, L. D., Bustamante, C. D., Evans, J. P., Landrum, M. J., & Watson, M. S. (2018). ClinGen — the clinical genome resource. New England Journal of Medicine, 372(23), 2235–2242. https://doi.org/10.1056/NEJMsr1406261
Our Key Guarantees
- ✓ 100% Plagiarism-Free
- ✓ On-Time Delivery
- ✓ Student-Friendly Pricing
- ✓ Human-Written Papers
- ✓ Free Revisions (14 days)
- ✓ 24/7 Live Support